









G6PC
The G6PC gene encodes glucose-6-phosphatase, a key enzyme that catalyzes the final step of gluconeogenesis and glycogenolysis, enabling the release of free glucose into the bloodstream during fasting.
Mutations in G6PC cause glycogen storage disease type Ia (GSD Ia), a rare metabolic disorder characterized by fasting hypoglycemia, hepatomegaly, lactic acidosis, and other systemic complications.
What is G6PC (Glucose-6-Phosphatase Catalytic Subunit)?
The G6PC gene, located on chromosome 17q21.31, encodes the enzyme glucose-6-phosphatase (G6Pase-α). It catalyzes the hydrolysis of glucose-6-phosphate (G6P) into free glucose and phosphate, the final and rate-limiting step of gluconeogenesis and glycogenolysis.
This process is essential for maintaining euglycemia, particularly during fasting.
The enzyme is embedded in the endoplasmic reticulum (ER) membrane, where it partners with the glucose-6-phosphate translocase (SLC37A4 gene product) to enable glucose production.
Role in Glucose Metabolism
G6PC is essential for maintaining blood glucose during fasting by facilitating the terminal step of:
- Gluconeogenesis: Synthesis of glucose from non-carbohydrate sources
- Glycogenolysis: Breakdown of glycogen to glucose
Tissue Expression
The G6PC protein is highly expressed in:
- Liver (major regulator of glucose production)
- Kidney
- Intestines
When is G6PC Enzyme Activity or Genetic Testing Relevant?
G6PC genetic testing should be considered in the following situations:
Suspected Glycogen Storage Disease Type Ia (GSD Ia)
G6PC testing is indicated when patients present with clinical features suggestive of GSD Ia, including fasting hypoglycemia, hepatomegaly, lactic acidosis, hyperlipidemia, and growth retardation.
These findings result from impaired glucose production and glycogen accumulation due to deficient glucose-6-phosphatase activity.
Positive Family History of GSD Ia
Genetic testing is recommended for individuals with a family history of GSD Ia to identify carriers or at-risk individuals and to provide information for reproductive planning, prenatal diagnosis, or early diagnosis of affected family members.
Enzyme Activity Assay (Liver Biopsy)
Although historically used to confirm G6PC deficiency, direct measurement of glucose-6-phosphatase activity via liver biopsy has largely been replaced by less invasive molecular genetic testing.
However, it may still be considered in rare situations where genetic testing is inconclusive.
Differential Diagnosis of GSD Ib
G6PC testing is important to differentiate GSD Ia from Glycogen Storage Disease Type Ib (GSD Ib), which shares similar metabolic abnormalities but includes additional features such as neutropenia, recurrent infections, and inflammatory bowel disease.
It is essential to distinguish between GSD Ia and GSD Ib as it impacts both management and prognosis.
What Do Specific G6PC Mutations Mean?
Specific G6PC mutations may have the following significance:
Genetic Basis
GSD Ia is inherited in an autosomal recessive pattern. Over 85 mutations have been identified, and mutations frequently show ethnic clustering.
Common Pathogenic Mutations (Ethnicity-Specific Patterns)
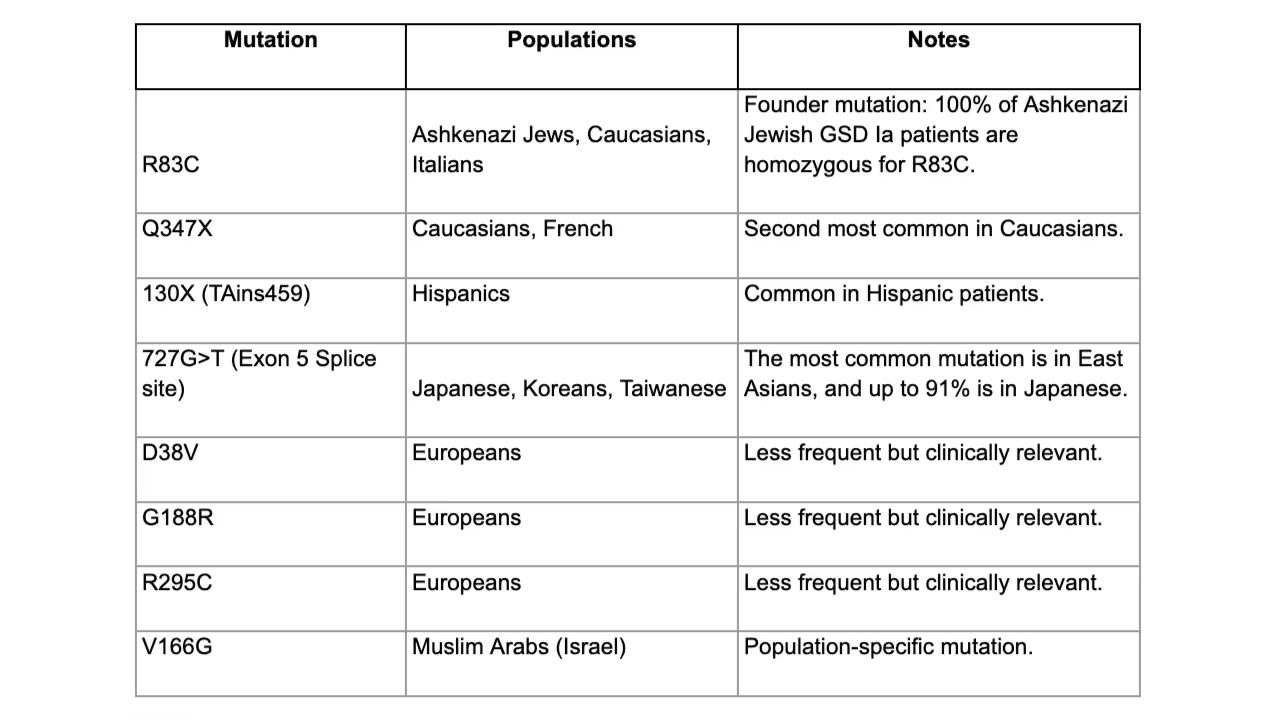
Pathophysiology
The pathophysiology of G6PC mutations may involve loss of function. In some cases, overexpression may also occur.
Loss of G6PC Function
Loss of G6PC function impairs the conversion of glucose-6-phosphate (G6P) into free glucose, leading to the cytoplasmic accumulation of G6P. This excess G6P is diverted into alternative metabolic pathways, resulting in the overproduction and storage of glycogen and lipids within cells.
The excessive buildup of these substances leads to hepatomegaly (enlarged liver) and nephromegaly (enlarged kidneys) due to cellular distension and organ dysfunction.
The inability to release free glucose into circulation causes fasting hypoglycemia, as the body cannot maintain blood glucose levels during periods of fasting. Additionally, excess G6P enhances glycolysis and lactate production, leading to lactic acidosis.
The shunting of metabolic intermediates into lipid synthesis contributes to hyperlipidemia, while disturbances in purine metabolism due to the altered energy balance result in hyperuricemia.
Over time, these metabolic derangements impair growth, contributing to growth retardation, a common feature of Glycogen Storage Disease Type Ia (GSD Ia).
Overexpression of G6PC
In some cases, particularly in altered glucose management, overexpression of G6PC may occur.
In diabetes, insulin resistance or low insulin levels fail to properly suppress G6PC expression, causing the liver to produce too much glucose. This increase in G6PC activity contributes to the high blood sugar levels commonly seen in people with diabetes.
Genotype-Phenotype Correlations
Certain G6PC mutations have been associated with variability in disease severity and clinical manifestations.
Thus, while genotyping provides valuable information, it does not always reliably predict the severity of symptoms or long-term complications.
Diagnostic Considerations
G6PC genetic testing has largely replaced invasive liver biopsy and enzyme assays and definitively diagnoses GSD Ia. Genetic testing is essential for confirming diagnosis, enabling prenatal testing, carrier screening, and guiding genetic counseling.
Broad next-generation sequencing (NGS) is recommended for individuals of mixed or unknown ancestry.
Management of GSD
Management of GSD Ia relies on lifelong dietary therapy to prevent hypoglycemia and minimize metabolic complications. Treatment may include continuous nocturnal glucose infusion in infants, uncooked cornstarch for older children, and a vegetarian diet.
Long-term management addresses risks such as hepatic adenomas (with about a 10% malignancy risk), gout, renal disease, osteoporosis, growth delay, and hepatocellular carcinoma.
Emerging therapies, including gene therapy and enzyme replacement, are under investigation. Animal studies suggest that restoring as little as 7% of normal G6PC activity may prevent hypoglycemia, though higher activity may be needed to protect renal function.
Test Procedure and Interpretation
Testing for G6PC is performed as a genetic test to look for mutations in the gene that would alter functional protein availability. The following section outlines the testing procedures and interpretation.
Testing Procedure and Preparation
Genetic testing involves blood, saliva, or cheek swab samples, although specialized laboratories may recommend different sample types, including chorionic villus sampling.
A cheek swab or saliva sample is easily obtained from the comfort of home, while blood samples typically require a blood draw.
Normal Reference Ranges
Normal reference ranges for G6PC genetic testing are considered to be without mutations that can alter the activity of the G6PC proteins.
Clinical Implications of Positive G6PC Mutations
The clinical implications of a positive G6PC mutation test result will vary by individual, although G6PC mutations in symptomatic patients may signal a need for further assessment and possibly treatment, especially in the setting of symptoms of impaired glucose metabolism, especially in infants or young children.
Patients or practitioners with questions about the clinical implications of G6PC mutations should seek further assessment with a genetic counselor or expert.
What Does the Absence of Pathogenic G6PC Mutations Mean?
The absence of mutations in the G6PC gene does not exclude other causes of hypoglycemia or hepatomegaly. If clinical suspicion persists, further metabolic or genetic evaluation is warranted.
Consider testing for SLC37A4 mutations if neutropenia or myeloid dysfunction is present (these symptoms suggest GSD Ib).
Notes for Clinicians
- GSD Ia results from defective gluconeogenesis and glycogenolysis → fasting intolerance.
- Genetic testing is essential for:
- Confirming diagnosis
- Differentiating GSD Ia from GSD Ib
- Guiding management and genetic counseling
- Carrier screening is important in high-risk populations.
- Elevated G6PC expression is also implicated in diabetes due to its role in hepatic glucose production.
Order Genetic Testing
Click here to compare genetic test panels and order genetic testing for health-related SNPs.




Chang, C.; Chen, C. Sep-Oct 2014 - Volume 34 - Issue 5 : Journal of Medical Sciences. (2025). Lww.com. https://journals.lww.com/joms/fulltext/2014/34050/unique_glucose_6_phosphatase
Chou JY, Mansfield BC. Mutations in the glucose-6-phosphatase-alpha (G6PC) gene that cause type Ia glycogen storage disease. Hum Mutat. 2008 Jul;29(7):921-30. doi: 10.1002/humu.20772. PMID: 18449899; PMCID: PMC2475600.
G6PC gene: MedlinePlus Genetics. (n.d.). Medlineplus.gov. https://medlineplus.gov/genetics/gene/g6pc/
G6PC1 glucose-6-phosphatase catalytic subunit 1 [Homo sapiens (human)] - Gene - NCBI. (2024). Nih.gov. https://www.ncbi.nlm.nih.gov/gene/2538
Hutton, J. C., & O’Brien, R. M. (2009). Glucose-6-phosphatase Catalytic Subunit Gene Family. Journal of Biological Chemistry, 284(43), 29241–29245. https://doi.org/10.1074/jbc.r109.025544
Lei KJ, Chen YT, Chen H, Wong LJ, Liu JL, McConkie-Rosell A, Van Hove JL, Ou HC, Yeh NJ, Pan LY, et al. Genetic basis of glycogen storage disease type 1a: prevalent mutations at the glucose-6-phosphatase locus. Am J Hum Genet. 1995 Oct;57(4):766-71. PMID: 7573034; PMCID: PMC1801521.
Melkonian EA, Asuka E, Schury MP. Physiology, Gluconeogenesis. [Updated 2023 Nov 13]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK541119/
Patino SC, Orrick JA. Biochemistry - Glycogenolysis. [Updated 2024 Jan 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK549820/
Test for
G6PC






